
Anwendungsbeispiele
Eine Auswahl an Beispielen aus verschiedenen Instituten am Helmholtz-Zentrum Hereon, die derzeit auf dem Hochleistungsrechner "Strand" laufen:
Die Abteilung Hydrodynamik und Datenassimilation führt numerische Simulationen der 3-D-Ozean-Zirkulation und von Meereswellen durch. Die verwendeten Methoden sind numerische Modellierung, Datenanalyse und Datenassimilation. Ein Schwerpunkt liegt auf der Datenassimilation (für z.B. SST, SSS, Hs), d.h. der Synergie zwischen Modell und Beobachtungen (in-situ, Satellit, HF-Radar), um den Endnutzern zuverlässige Produkte zur Verfügung zu stellen.
Eine weitere wichtige Aktivität ist die Integration verschiedener Modelle und die Implementierung von biogeochemischen Kreisläufen im Ozean. Bei der Integration verschiedener Modelle gilt ein Kopplungskonzept: Wechselwirkungsprozesse zwischen Wind und Wellen, d.h. Atmosphäre und Ozean werden durch separate Modelle berechnet und tauschen über einen Koppler Informationen an den Modellrändern aus. Diese Kopplung ermöglicht es den Wissenschaftlern, nichtlineare Rückkopplungen zwischen den Komponenten des Erdsystems zu untersuchen und ist von besonderem Forschungsinteresse.
All diese Aufgaben erfordern ein hohes Maß an Rechenleistung und Festplattenplatz. Hier kommt der HPC-Cluster "Strand" am Helmholtz-Zentrum Hereon ins Blickfeld und ermöglicht es den Wissenschaftlern, ihre Forschungsaufgaben effizient zu erledigen.
Neben der Forschung bietet die Abteilung auch Unterstützung für das operationelle System und den Betrieb von präoperativen Systemen (COSYNA, BSH, DWD, Deutsche Marine, CMEMS).
Luftqualität hat einen hohen Einfluss auf die Gesundheit. Schadstoffe wie NO2 oder Feinstaub beeinträchtigen die Atemwege von Mensch und Tier und können zu einer verkürzten Lebensdauer führen. In der Abteilung Chemietransportmodellierung untersuchen Wissenschaftler Luftschadstoffe aus verschiedenen Quellen, ihren Transport und die chemische Umwandlung in der Luft, in der wir leben. Fragen wie "Welchen Beitrag leistet die Schadstoffquelle X zu den Schadstoffkonzentrationen in der Region A?" oder "Was wäre, wenn sich etwas ändern würde (z.B. Emissionen, Klima, Politik, Vorschriften)?" werden mit Hilfe von numerischen Chemietransportmodellen beantwortet.
Die Abteilung "Chemietransportmodellierung" am Helmholtz-Zentrum Hereon hat einen Schwerpunkt in der Abschätzung und Modellierung von Schiffsemissionen und den entsprechenden Luftkonzentrationen im regionalen Maßstab. In verschiedenen Projekten wurden Szenarien mit unterschiedlichen Gesetzgebungen berechnet.
Ein weiteres zentrales Thema der Abteilung ist die Modellierung von Verkehr und Chemie im Stadtmaßstab. Da Städte sehr komplexe Strukturen aufweisen, lassen sich Luftströmung und Schadstoffausbreitung weniger leicht vorhersagen als auf regionaler Ebene. Im Zusammenhang mit den Emissionen des Schiffsverkehrs und der Luftverschmutzung sind die Hafenstädte von besonderem Forschungsinteresse in der Gruppe. Neben der Berechnung von Konzentrationen entwickelt die Abteilung ein Emissionsmodell namens HiMEMO.
All diese Aufgaben verbrauchen auf HPC-Clustern eine bemerkenswerte Menge an CPU-Zeit und erfordern massive Datenspeicherkapazitäten. Hier stellt der Cluster "Strand" die für diese Berechnungen erforderliche Rechenleistung und Infrastruktur zur Verfügung.
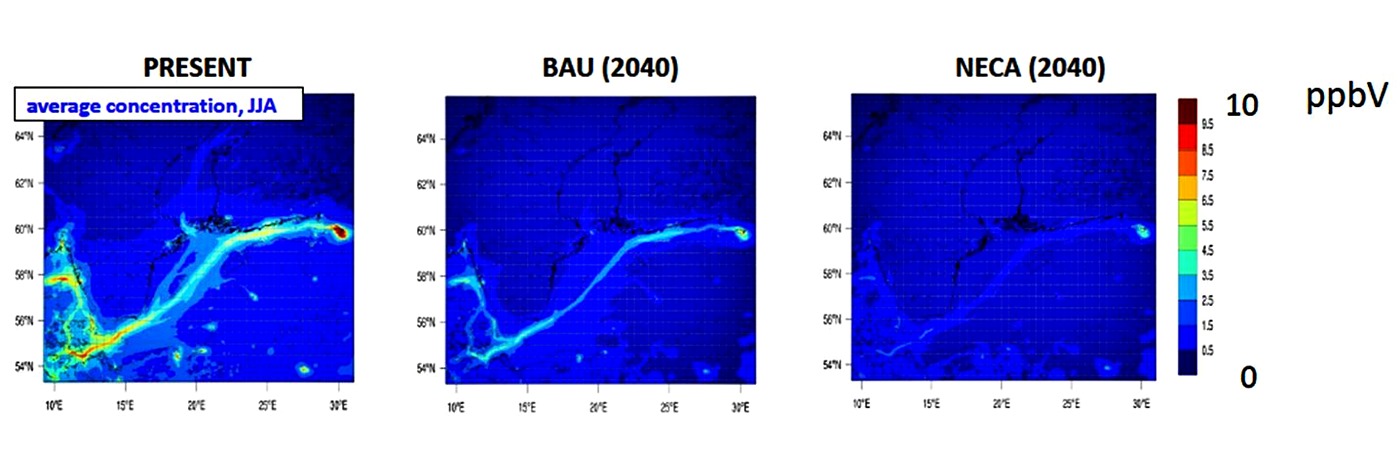
(Grafik: Hereon)
Erforschung von grundlegenden Mechanismen der Magnesiumkorrosion durch atomistische Simulationen
Da Experimente nicht immer ausreichen, um die komplexen grundlegenden Prozesse der Magnesiumkorrosion einschlägig zu beschreiben und zu verstehen, bieten atomistische Simulationen eine attraktive Möglichkeit, um tiefere Einblicke auf atomarer Ebene zu gewähren. Die dafür gewählten Methoden basieren dabei entweder - wie im Rahmen der klassischen Molekulardynamik - auf empirischen Kraftfeldern, oder nutzen ab initio Methoden wie die Dichtefunktionaltheorie, welche die direkte Berechnung der elektronischen Struktur erlaubt. Auf diese Weise ist es möglich, Reaktionsbarrieren zu identifizieren, Ladungsverschiebungen zu beschreiben und generell ein Verständnis für die energetischen Einflüsse innerhalb der untersuchten Reaktionsmechanismen zu gewinnen. Mithilfe der Molekulardynamik ist es darüberhinaus möglich, Reaktionskinetiken zu bestimmen und beispielsweise das Degradationsverhalten von Magnesium zu analyisieren.
Das High-Performance-Computing (HPC) ermöglicht hierbei den Wissenschaftlern der Abteilung Atomistische Simulationen die Simulation von großen Systemen (mehrere Hundert bis mehrere Tausend Atome) sowie die Durchführung von Hochdurchsatzansätzen zur maximierten Datenerhebung für Anwendungen des maschinellen Lernens. So können zum Beispiel energetisch optimierte molekulare Strukturen mit experimentellen sowie Ergebnissen aus atomistischen Simulationen kombiniert werden, um etwaige Struktur-Eigenschafts-Beziehungen zu identifizieren.
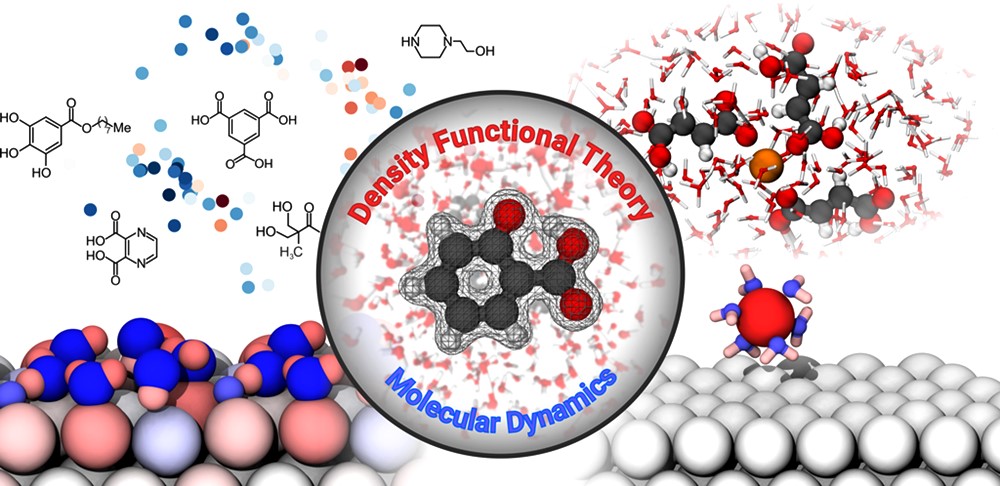
Methoden der atomistischen Materialmodellierung bieten die Möglichkeit, elementare Korrosionsprozesse auf einem atomaren Level abzubilden. (von links oben im Uhrzeigersinn) Landkarte der Korrosionsinhibitoren basierend auf molekularen Ähnlichkeiten; Komplexierung eines Magnesiumions durch Fumarsäure; ein Magnesiumion geht in Lösung; Oberflächenladungen in der Magnesium-Wasser-Grenzschicht. (Grafik: Hereon)
Untersuchung der Struktur-Eigenschafts-Beziehungen nanoporöser Metalle
Das Verständnis der Struktur-Eigenschafts-Beziehung von offenporigen Materialien wie nanoporösem Gold erfordert eine detaillierte Kenntis der morphologischen und topologischen Details ihrer Mikrostruktur. Etablierte Modelle wie z.B. das Gibson-Ashby-Skalierungsgesetz, die sich ausschließlich auf vergleichsweise einfach zu bestimmende morphologische Parameter wie das Porenvolumen stützen, erwiesen sind folglich nur in gewissen Grenzen fähig, Aussagen über das makroskopische Materialverhalten nanoporöser Metalle zu machen. Doch selbst anhand detaillierter Tomographien solcher Mikrostrukturen ist es schwierig, die relevanten Deskriptoren zu identifizieren, die das Ligamentnetzwerk adäquat im Sinne der benötigten Eingabeparameter für eine Struktur-Eigenschafts-Beziehung repräsentieren.
Um den Einfluss der morphologischen und topologischen Merkmale wie Ligamentgröße, Ligamentform und Verbindungen im Ligamentnetzwerk auf die makroskopische Konstitutivantwort von nanoporösem Metallen zu untersuchen, führen Wissenschaftler des Instituts für Werkstoffmechanik systematische numerische Studien mit verschiedenen Diskretisierungsansätzen (z.B. Voxel- oder Balkendiskretisierungen) von Diamant-Einheitszellen durch. Im Gegensatz zu Experimenten erlauben solche numerischen Studien eine systematische Variation einzelner mikrostruktureller Merkmale und helfen so deren Einfluss auf die makroskopischen Eigenschaften zu isolieren und letztlich die Mikrostrukturdeskriptoren zu ermitteln, aus denen geeignete Struktur-Eigenschafts-Beziehungen abgeleitet werden können.
Ausgehend von perfekt geordneten Strukturen kann die Komplexität sukzessive durch Hinzufügen von Variationen in der Ligamentform, struktureller Unordnung und Veränderung der Konnektivität erhöht werden. Während dadurch reale Mikrostrukturen schrittweise angenähert werden, kann gleichzeitig der Einfluss der individuellen morphologischen Parameter auf die makroskopischen Eigenschaften im Sinne von Elastizität, Plastizität und Funktionalität erfasst werden. Auf der Grundlage dieser Daten ergibt sich ein klares Bild darüber, welche Deskriptoren wesentlich für die Vorhersage welcher effektiven Eigenschaften sind.

Abbildung: Elastisch plastische Verformung eines nanoporösen Metalls, vorhergesagt durch FE-Modelle zunehmender Komplexität (Grafik: Hereon)